scRNA-seq与Bulk RNA-seq联合分析:从细胞图谱到临床预测
2025-12-09
相对于Bulk RNA测序,scRNA-seq(单细胞RNA测序)能够分离组织中的每一个细胞,分别进行测序,从而绘制出细胞图谱,鉴定新的细胞亚型,揭示细胞间的异质性,能发现稀有但关键的细胞群体。但是在转录组的覆盖广度与测序深度方面,单细胞RNA测序的数据通常难以达到批量RNA测序的水平,这主要归因于其在细胞分离、分子捕获与扩增环节中不可避免的技术损耗。在当今的组学研究中,单细胞RNA测序和Bulk RNA测序进行联合分析,巧妙地将二者结合,为生物学研究带来了更多的视角。
核心逻辑:优势互补,形成完整证据链
u scRNA-seq:在单细胞分辨率下深度发现,无偏倚地发现新的细胞亚群、关键基因、调控网络和细胞间通讯。
u Bulk RNA-seq:利用大量临床样本广泛验证,验证发现的普适性、临床相关性及预后价值,将机制转化为标志物。.
案例一:scRNA-seq和Bulk RNA-seq联合分析发现预测性生物标志物
参考文献:Liu H, Sima X, Xiao B, et al. Integrated analysis of single-cell and bulk rna sequencing data reveals a myeloid cell-related regulon predicting neoadjuvant immunotherapy response across cancers [J]. J Transl Med, 2024, 22(1): 486.
研究目标:找到能预测多种癌症对免疫检查点抑制剂响应的关键因子。
联合分析策略:
1. scRNA-seq发现:
对治疗响应/非响应的肺癌患者样本进行scRNA-seq分析,构建肿瘤微环境单细胞图谱。使用pySCENIC算法,挖掘基因调控网络,发现PPARG调控子在响应者髓系细胞中特异性高活性。
2. Bulk RNA-seq验证与转化:
将PPARG调控子的23个靶基因作为“特征基因集”。在TCGA泛癌数据集和5个独立免疫治疗队列中,通过GSVA算法为每个Bulk样本计算“PPARG活性得分”。
结果:该得分在多个队列中均能有效预测患者更好的免疫治疗响应和生存期。
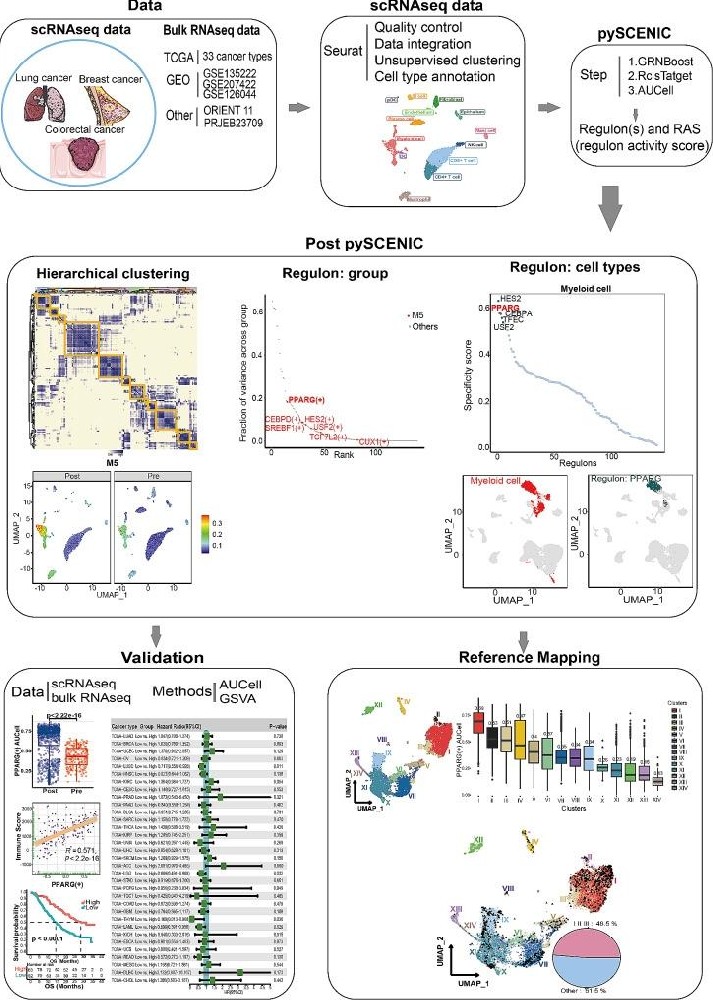
总结:scRNA-seq机制发现 → Bulk RNA-seq大队列临床验证,将单细胞发现转化为可在临床大样本中应用的量化评分。文章对极少数(n=2) 关键的治疗前后配对样本进行scRNA-seq,作为“发现引擎”,锁定关键因子PPARG调控子,然后,在已有的大量公共Bulk数据(如TCGA) 中进行验证。
案例二:scRNA-seq和Bulk RNA-seq联合分析解析M2细胞亚群
参考文献:He S, Li C, Lu M, et al. Comprehensive analysis of scrna-seq and bulk rna-seq reveals the non-cardiomyocytes heterogeneity and novel cell populations in dilated cardiomyopathy [J]. J Transl Med, 2025, 23(1): 17.
研究目标:揭示扩张型心肌病中非心肌细胞的异质性及其在疾病中的作用。
联合分析策略:
1. scRNA-seq发现:
整合DCM患者和正常人的心脏scRNA-seq数据,绘制了详尽的非心肌细胞图谱。深入分析成纤维细胞和髓系细胞的异质性,鉴定出两个功能迥异的M2巨噬细胞新亚群:M2-like1和M2-like2。并进行了细胞轨迹、代谢、转录调控和细胞通讯等深度分析。
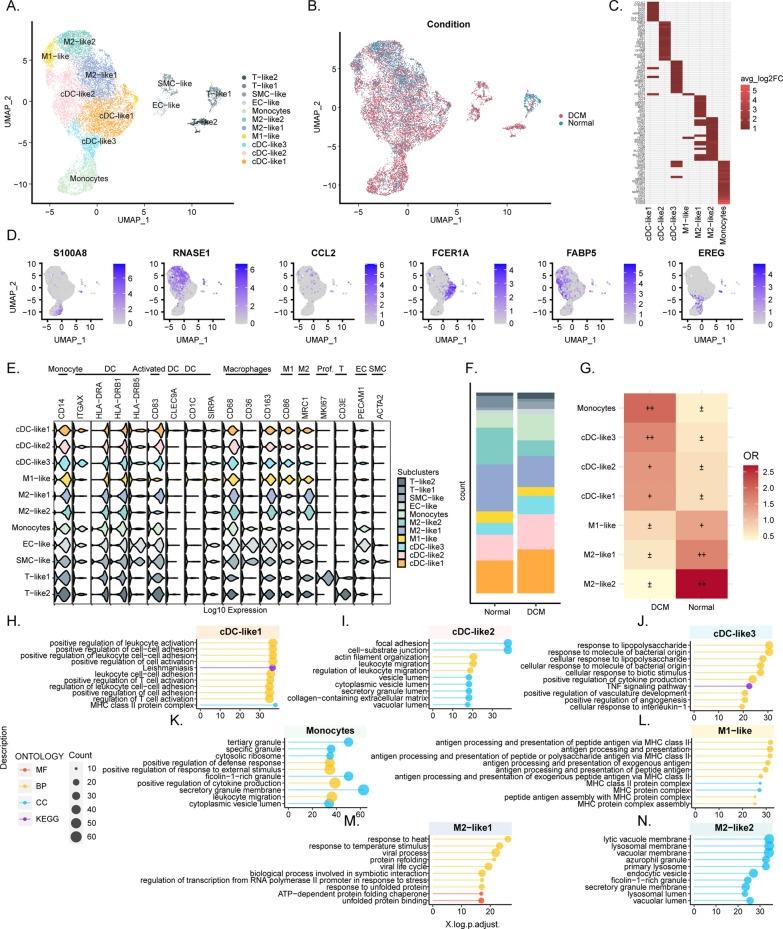
2. Bulk RNA-seq验证与量化:
为了验证scRNA-seq发现的细胞比例变化,研究者使用了多种反卷积算法,在Bulk数据中估算不同细胞类型的丰度。他们利用ssGSEA计算M2-like2特征基因集的富集分数,在大队列时数据中证实M2-like2巨噬细胞在DCM患者中显著富集。
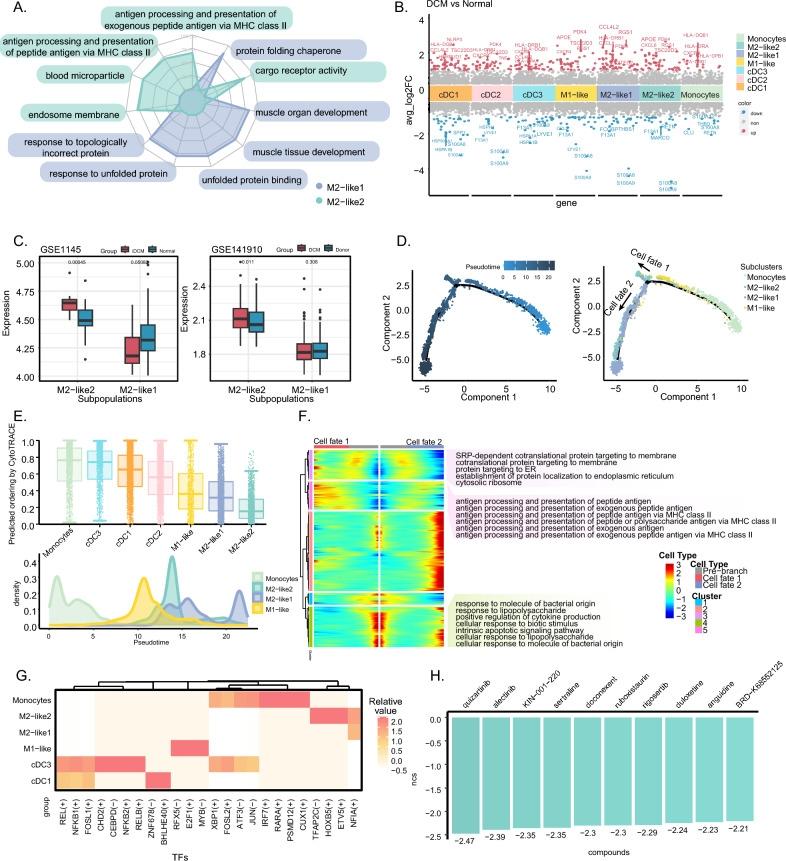
总结:scRNA-seq图谱解析 → Bulk RNA-seq细胞比例验证,整合10个样本的单细胞数据构建核心图谱,描绘组织的核心细胞图谱,鉴定出新的M2巨噬细胞亚群。随后,通过对322个Bulk样本的反卷积分析进行验证。
案例三:多组学联合解析肿瘤异质性与细胞互作
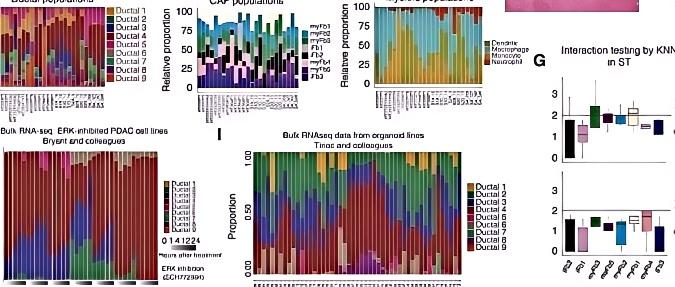
参考文献:Loveless IM, Kemp SB, Hartway KM, et al. Human pancreatic cancer single-cell atlas reveals association of cxcl10+ fibroblasts and basal subtype tumor cells [J]. Clin Cancer Res, 2025, 31(4): 756-772.
研究目标:揭示胰腺癌的细胞异质性,并找出与不良预后相关的肿瘤细胞亚型及其独特的肿瘤微环境。
联合分析策略:
1. scRNA-seq发现:
整合229例人胰腺样本的scRNA-seq数据(超70万细胞),构建了全面的胰腺癌单细胞图谱。通过单细胞图谱鉴定出9个肿瘤上皮细胞亚群,其中两个(Ductal 4, Ductal 7)为基底样亚型,与患者预后不良显著相关。发现一类CXCL10+ 肌成纤维细胞 与基底样肿瘤细胞在分析中存在关联。
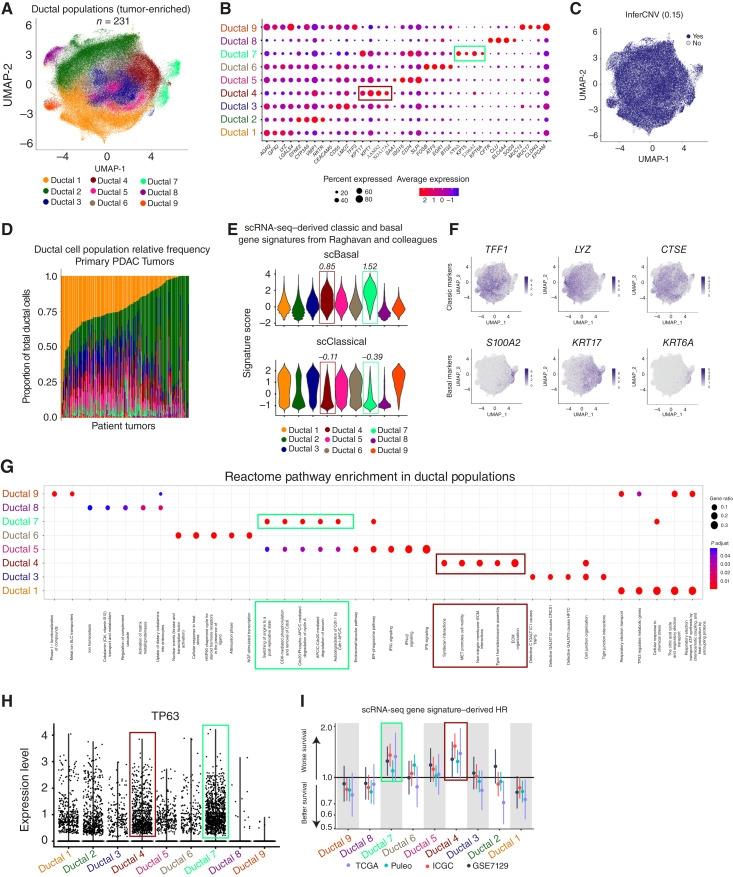
2. Bulk RNA-seq验证与关联:
将9个细胞亚群的基因特征在744例Bulk RNA-seq样本中进行反卷积分析。验证基底样亚型(Ductal 4/7)的基因特征确实与患者的总生存期缩短显著相关。
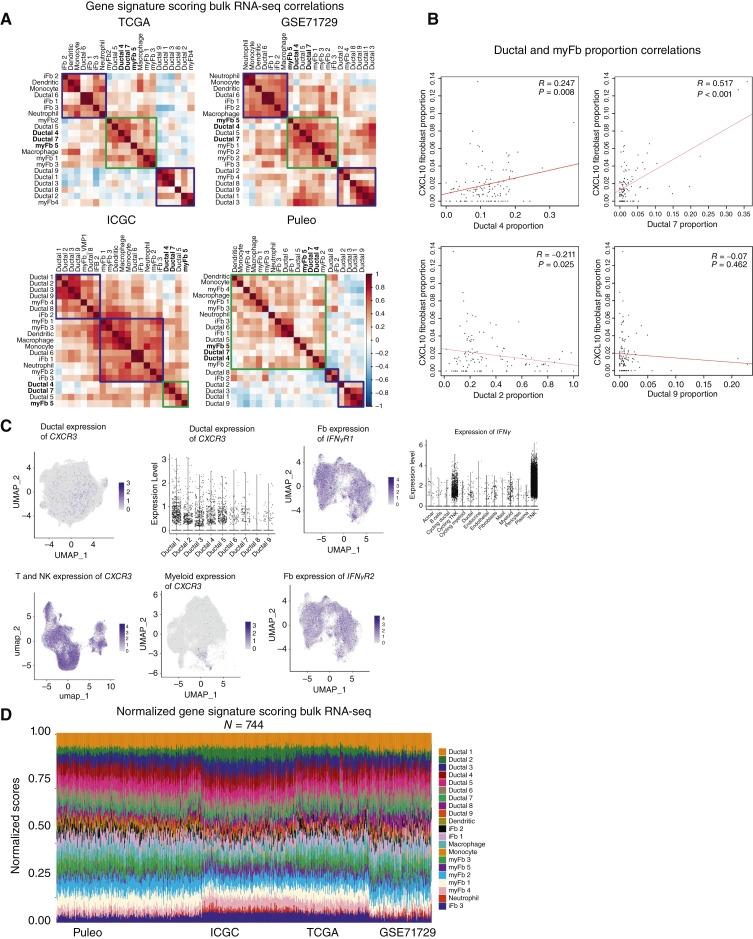
3. 空间转录学原位验证:
利用22个空间转录组样本进行验证,证实了Ductal 4(基底样)肿瘤细胞与CXCL10+ 成纤维细胞在空间上紧密相邻。
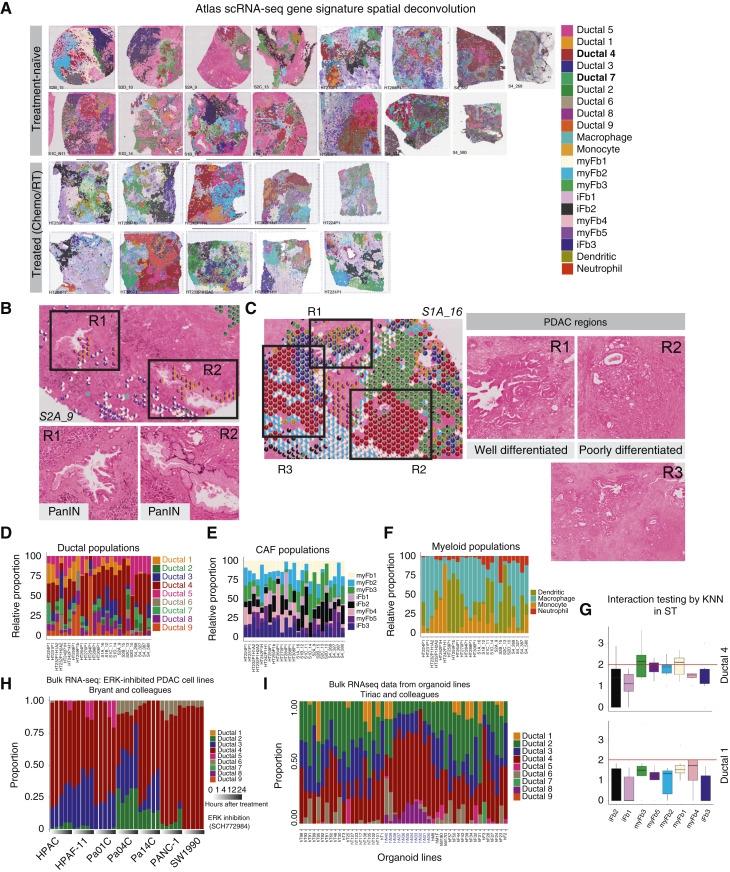
总结:scRNA-seq图谱构建 → Bulk RNA-seq生存关联与细胞互作推测 → 空间技术原位证实。文章通过聚合12项研究中229例样本的scRNA-seq,以及整理数据库中744例Bulk RNA-seq,并通过22 个PDAC组织切片的空间转录组进行原位验证;构建权威细胞图谱、并完成多组学验证的完整研究,通常涉及数百个样本和多技术平台整合,是大型、系统性的投入。
这三篇研究清晰地展示了,根据不同的生物学问题,scRNA-seq和Bulk RNA-seq可以灵活地以不同方式组合。scRNA-seq提供分辨率,Bulk RNA-seq提供规模与临床关联,二者结合是实现“从机制到临床”转化的关键方法。三篇案例组学检测的样本各不相同,合理利用检测预算也十分重要。可将预算精准投入到由3-8个核心样本构成的scRNA-seq探索阶段,旨在完成高质量的细胞图谱绘制与关键靶标发现;随后由数十至数百个样本的Bulk RNA-seq验证阶段,在独立大队列中坚实验证发现的普适性与临床价值,双组学检测与联合分析环节的成本分别约为20万左右,确保实现研究价值的最大化。
 技术咨询:
技术咨询: